A Sequential Solution¶
Problem Definition¶
Working with actual ligand and protein data is beyond the scope of this example, so we will represent the computation by a simpler string-based comparison.
Specifically, we simplify the computation as follows:
- Proteins and ligands will be represented as (randomly-generated) character strings.
- The docking-problem computation will be represented by comparing a ligand string L to a protein string P. The score for a pair [L, P] will be the maximum number of matching characters among all possibilities when L is compared to P, moving from left to right, allowing possible insertions and deletions. For example, if L is the string “cxtbcrv” and P is the string “lcacxtqvivg,” then the score is 4, arising from this comparison of L to a segment of P:

This is not the only comparison of that ligand to that protein that yields four matching characters. Another one is
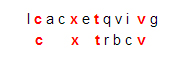
However, there is no comparison that matches five characters while moving from left to right, so the score is 4.
Implementation¶
The example program dd_serial.cpp provides a sequential C++ implementation of our simplified drug design problem.
Note
The program optionally accepts up to three command-line arguments:
- maximum length of the (randomly generated) ligand strings
- number of ligands generated
- protein string to which ligands will be compared
Compilation:¶
A straightforward compile can be used for this sequential example:
g++ -o dd_serial dd_serial.cpp
The Code¶
In this implementation, the class MR encapsulates the map-reduce steps Generate_tasks(), Map(), and Reduce() as private methods (member functions of the class), and a public method run() invokes those steps according to a map-reduce algorithmic strategy (see previous Introduction section for detailed explanation). We have highlighted calls to the methods representing map-reduce steps in the following code segment from MR::run().
Comments¶
We use the STL containers queue<> and vector<> to hold the results from each of the map-reduce steps: namely, the task queue of ligands to process, the list key-value pairs produced by the Map() phase, and the list of resulting key-value pairs produced by calls to Reduce(). We define those container variables as data members in the class MR:
queue<string> tasks;
vector<Pair> pairs, results;
Here, Pair is a struct representing key-value pairs with the desired types:
1 2 3 4 5
struct Pair { int key; string val; Pair(int k, const string &v) {key=k; val=v;} };
In the example code, Generate_tasks() merely produces nligands strings of random lower-case letters, each having a random length between 0 and max_ligand. The program stores those strings in a task queue named tasks.
For each ligand in the task queue, the Map() function computes the match score from comparing a string representing that ligand to a global string representing a target protein, using the simplified match-scoring algorithm described above. Map() then yields a key-value pair consisting of that score and that ligand, respectively.
The key-value pairs produced by all calls to Map() are sorted by key in order to group pairs with the same score. Then Reduce() is called once for each of those groups in order to yield a vector of Pairs consisting of a score s together with a list of all ligands whose best score was s.
Note
Map-reduce frameworks such as the open-source Hadoop commonly use sorting to group values for a given key, as does our program. This has the additional benefit of producing sorted results from the reduce stage. Also, the staged processes of performing all Map() calls before sorting and of performing all Reduce() calls after the completion of sorting are also common among map-reduce frameworks.
The methods Generate_tasks(), Map(), and Reduce() may seem like unnecessary complication for this problem since they abstract so little code. Indeed, we could certainly rewrite the program more simply and briefly without them. We chose this expression for several reasons:
- We can compare code segments from MR::run() directly with corresponding segments in upcoming parallel implementations to focus on the parallelization changes and hide the common code in method calls.
- The methods Generate_tasks(), Map(), and Reduce() make it obvious where to insert more realistic task generation, docking algorithm, etc., and where to change our map-reduce code examples for problems other than drug design.
- We use these three method names in descriptions of the map-reduce pattern elsewhere.
We have not attempted to implement the fault tolerance and scalability features of a production map-reduce framework such as Hadoop.
Questions for Exploration¶
- Compile and test run the sequential program. Determine values for the command-line arguments max_ligand``(maximum length of a ligand string) and ``nligands (total number of ligands to process) that lead to a tolerably long computation for experimenting (e.g., perhaps 15 seconds to a minute of computation). Note the following about our simplified computational problem:
- Our stand-in scoring algorithm is exponential in the lengths of the ligand and protein strings. Thus, a large value of max_ligand may cause an extremely lengthy computation. Altering max_ligand can help in finding a test computation of a desired order of magnitude.
- We expect the computation time to increase approximately linearly with the number of ligands nligands. However, if nligands is relatively small, you may notice irregular jumps to long computation times when increasing nligands. This is because our simple random algorithm for generating ligands produces ligand strings using random(), as well as ligands with random lengths as well as random content. Because of the order-of-magnitude effect of ligand length, a sudden long ligand (meaning more characters than those before) may greatly increase the computation time.
- If you have more realistic algorithms for docking and/or more realistic data for ligands and proteins, modify the program to incorporate those elements, and compare the results from your modified program to results obtained by other means (other software, wet-lab results, etc.).
